von Willebrand disease (VWD) is caused by quantitative and/or qualitative inherited abnormalities of von Willebrand factor (VWF). The prevalence of VWD associated with significant bleeding has been estimated to be approximately 1:10,000.
Quantitative Defects
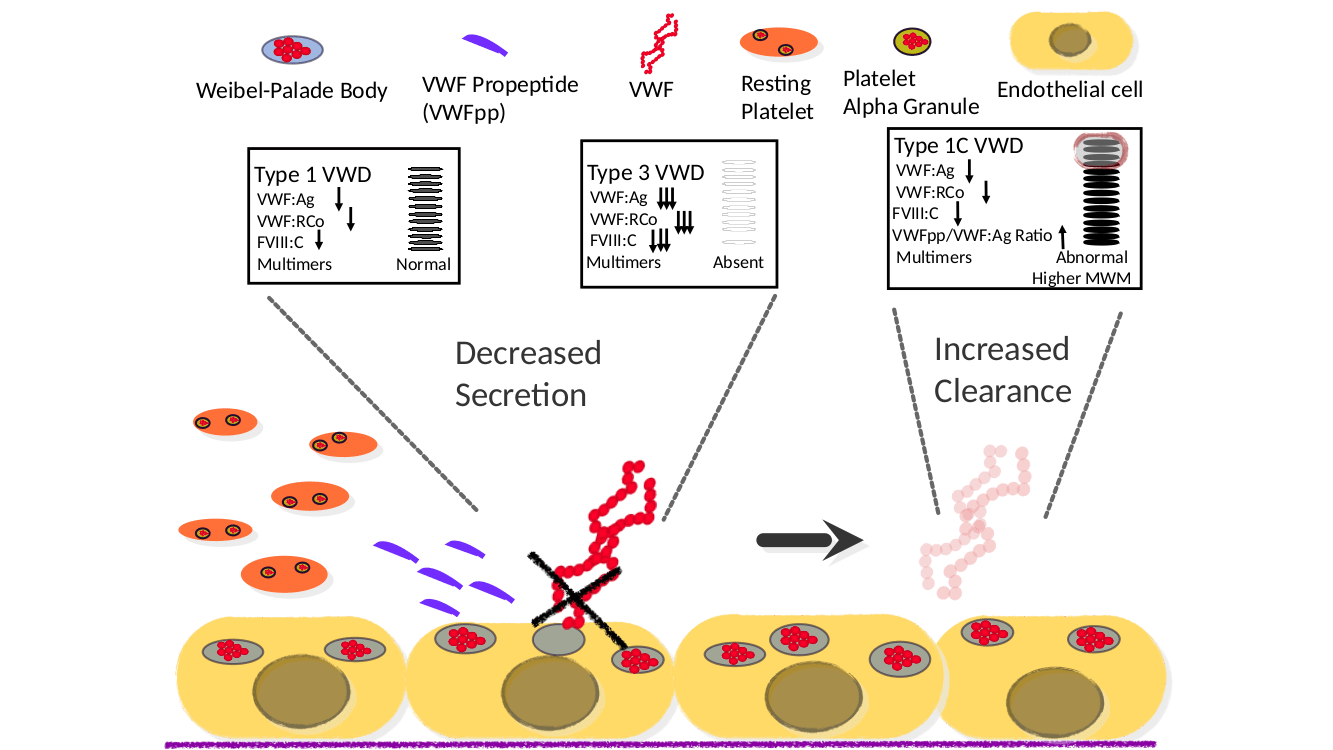
Type 1 VWD is the most commonly diagnosed VWD subtype, representing approximately 75% of all cases. In this subtype, VWF molecules are present in decreased amount in plasma, which are qualitatively normal. Mutations in VWF have been detected in approximately two thirds of cases when Type 1 VWD is defined by levels as low as < 30 IU/dL. In addition to the VWF gene itself, studies in our laboratory are also being undertaken to detect mutations at alternative genetic loci that contribute to VWF synthesis, secretion, and clearance and therefore likely affecting levels. A subset of type 1 patients has been designated type 1C, where the “C” indicates abnormally increased clearance of VWF from the circulation.
Type 3 VWD, is characterized by a severe absence of VWF due to deleterious mutations in VWF and whereas type 1 VWD is generally expressed in autosomal dominant fashion, type 3 is autosomal recessive.
Qualitative Abnormalities
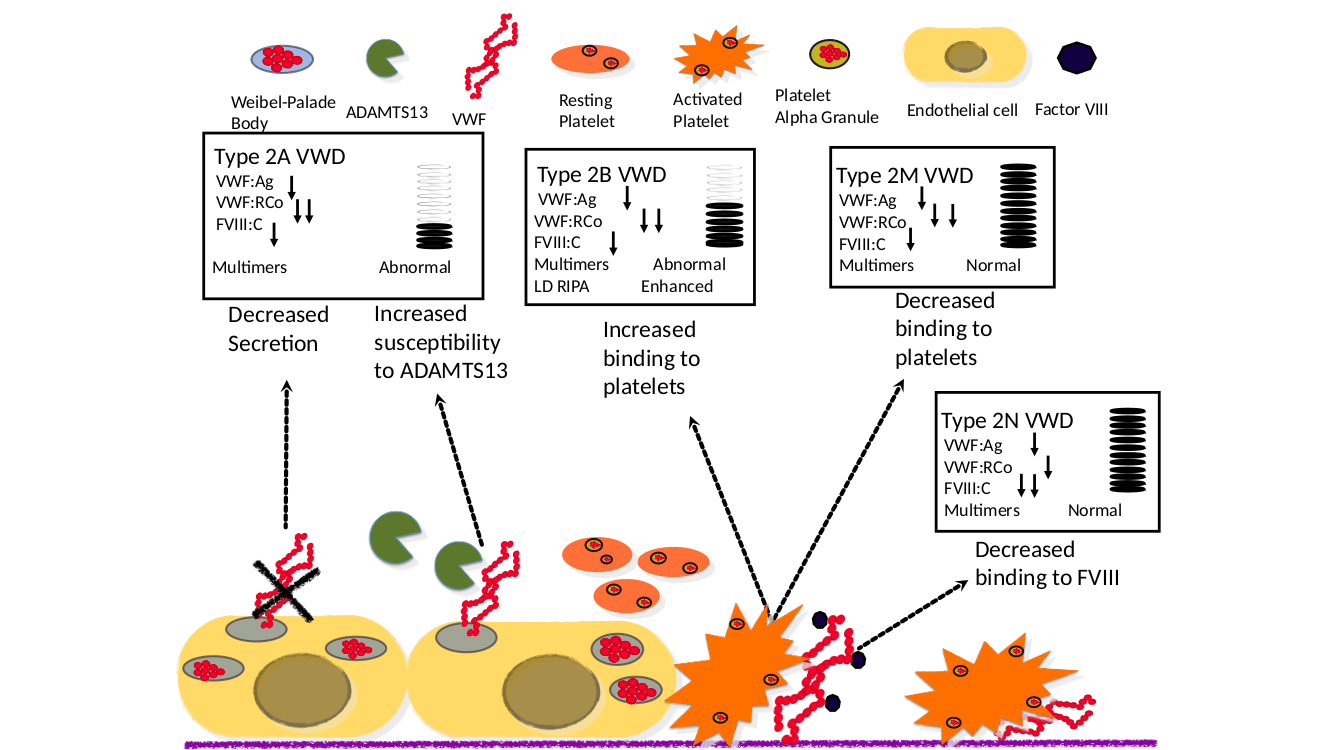
Patients with type 2 VWD subtypes have a qualitative abnormality of VWF. These subtypes are mostly defined by a defect in interactions with other ligands. In type 2A VWD, there is a lack of higher molecular weight multimers in plasma with corresponding loss of VWF function either caused by VWF mutations that prevent proper multimerization or by mutations that increase the susceptibility to proteolysis by ADAMTS13. In type 2B VWD, gain of function missense mutations within VWF increase the ability to bind the platelet receptor GP1bα leading often to a mild to moderate thrombocytopenia. Patients with type 2M VWD have mutations in the VWF platelet binding site, while patients with type 2N VWD (Normandy) exhibit mutations in the Factor VIII binding site.